Walkthrough on reading, visualizing, manipulating molecules and their properties for Cheminformatics-related tasks
Published
May 11, 2021
Walkthrough on reading, visualizing, manipulating molecules and their properties using Pandas and RDKit for Cheminformatics-related tasks. In this file we will be primarly using SMILES to describe the molecules of choice.
What are SMILES?
Simplified molecular input line entry system (SMILES) is a form of line notation for describing the structure of chemical species using text strings. More details can be found here
Other resources:
Nice low-barrier intro to using some basic functions in RDkit: Xinhao Lin’s RDKit Cheatsheet, I’ve adopted some of the functions frrom that blog in here:
Compilation of various recipes submitted by the community: * RDkit Cookbook
Install necessary modules
# collapse_output# Install requirements for the tutorial!pip install pandas rdkit-pypi mols2grid matplotlib scikit-learn ipywidgets
Requirement already satisfied: pandas in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (1.3.4)
Requirement already satisfied: rdkit-pypi in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (2021.9.2.1)
Requirement already satisfied: mols2grid in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (0.1.0)
Requirement already satisfied: matplotlib in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (3.5.1)
Requirement already satisfied: scikit-learn in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (1.0.2)
Requirement already satisfied: pytz>=2017.3 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from pandas) (2021.3)
Requirement already satisfied: python-dateutil>=2.7.3 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from pandas) (2.8.2)
Requirement already satisfied: numpy>=1.21.0 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from pandas) (1.21.4)
Requirement already satisfied: jinja2 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from mols2grid) (3.0.3)
Requirement already satisfied: pillow>=6.2.0 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from matplotlib) (8.4.0)
Requirement already satisfied: cycler>=0.10 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from matplotlib) (0.11.0)
Requirement already satisfied: pyparsing>=2.2.1 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from matplotlib) (3.0.6)
Requirement already satisfied: fonttools>=4.22.0 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from matplotlib) (4.28.3)
Requirement already satisfied: kiwisolver>=1.0.1 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from matplotlib) (1.3.2)
Requirement already satisfied: packaging>=20.0 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from matplotlib) (21.3)
Requirement already satisfied: joblib>=0.11 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from scikit-learn) (1.1.0)
Requirement already satisfied: scipy>=1.1.0 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from scikit-learn) (1.7.3)
Requirement already satisfied: threadpoolctl>=2.0.0 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from scikit-learn) (3.1.0)
Requirement already satisfied: six>=1.5 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from python-dateutil>=2.7.3->pandas) (1.16.0)
Requirement already satisfied: MarkupSafe>=2.0 in /Users/pghaneka/miniconda3/envs/small_mols/lib/python3.10/site-packages (from jinja2->mols2grid) (2.0.1)
import os import pandas as pdimport numpy as np
The majority of the basic molecular functionality is found in module rdkit.Chem
# RDkit importsimport rdkitfrom rdkit import Chem #This gives us most of RDkits's functionalityfrom rdkit.Chem import Drawfrom rdkit.Chem.Draw import IPythonConsole #Needed to show moleculesIPythonConsole.ipython_useSVG=True#SVG's tend to look nicer than the png counterpartsprint(rdkit.__version__)# Mute all errors except criticalChem.WrapLogs()lg = rdkit.RDLogger.logger() lg.setLevel(rdkit.RDLogger.CRITICAL)
2021.09.2
#----- PLOTTING PARAMS ----# import matplotlib.pyplot as pltfrom matplotlib.pyplot import cm# High DPI rendering for mac%config InlineBackend.figure_format ='retina'# Plot matplotlib plots with white background: %config InlineBackend.print_figure_kwargs={'facecolor' : "w"}plot_params = {'font.size' : 15,'axes.titlesize' : 15,'axes.labelsize' : 15,'axes.labelweight' : 'bold','xtick.labelsize' : 12,'ytick.labelsize' : 12,}plt.rcParams.update(plot_params)
Basics
# Beneze molecule using SMILES representationmol = Chem.MolFromSmiles("c1ccccc1")
mol is a special type of RDkit object
type(mol)
rdkit.Chem.rdchem.Mol
To display the molecule:
mol
By default SMILES (atleast the ones we deal with RDkit) accounts H atoms connected with C atoms implicitly based on the valence of the bond. You can add H explicitly using the command below:
Chem.AddHs(mol)
Chem.RemoveHs(mol)
# Convenience function to get atom indexdef mol_with_atom_index(mol):for atom in mol.GetAtoms(): atom.SetAtomMapNum(atom.GetIdx())return mol
# With atom indexmol_with_atom_index(mol)
# Another option using in-built index for atoms IPythonConsole.drawOptions.addAtomIndices =Truemol = Chem.MolFromSmiles("c1ccccc1")mol
Descriptors for molecules
We can find more information about the molecule using rdkit.Chem.Descriptors : More information here
from rdkit.Chem import Descriptors
# To get molecular weightmol_wt = Descriptors.ExactMolWt(mol)print('Mol Wt: {:0.3f}'.format(mol_wt))
Mol Wt: 78.047
# To get heavy atom weight, this ignore H atoms heavy_mol_wt = Descriptors.HeavyAtomMolWt(mol)print('Heavy Mol Wt: {:0.3f}'.format(heavy_mol_wt))
Heavy Mol Wt: 72.066
# To get number of rings ring_count = Descriptors.RingCount(mol)print('Number of rings: {:0.3f}'.format(ring_count))
Number of rings: 1.000
# To get number of rotational bonds rotatable_bonds = Descriptors.NumRotatableBonds(mol)print('Number of rotatable bonds: {:0.3f}'.format(rotatable_bonds))
Number of rotatable bonds: 0.000
# To get number of rings num_aromatic_rings = Descriptors.NumAromaticRings(mol)print('Number of aromatic rings: {:0.3f}'.format(num_aromatic_rings))
Number of aromatic rings: 1.000
Get some more information about the molecule:
MolToMolBlock: To get Coordinates and bonding details for the molecule. More details on this file type can be found here
Let’s look at the the C=O and the -NH species in the molecule
# Code from : https://www.rdkit.org/docs/GettingStartedInPython.html?highlight=maccs#drawing-moleculessub_pattern = Chem.MolFromSmarts('O=CCccN')hit_ats =list(diclofenac.GetSubstructMatch(sub_pattern))hit_bonds = []for bond in sub_pattern.GetBonds(): aid1 = hit_ats[bond.GetBeginAtomIdx()] aid2 = hit_ats[bond.GetEndAtomIdx()] hit_bonds.append( diclofenac.GetBondBetweenAtoms(aid1, aid2).GetIdx() )
d2d = rdMolDraw2D.MolDraw2DSVG(400, 400) # or MolDraw2DCairo to get PNGsrdMolDraw2D.PrepareAndDrawMolecule(d2d, diclofenac, highlightAtoms=hit_ats, highlightBonds=hit_bonds)d2d.FinishDrawing()SVG(d2d.GetDrawingText())
Specify individual color and bonds
rings = diclofenac.GetRingInfo()type(rings)
rdkit.Chem.rdchem.RingInfo
# Code from: http://rdkit.blogspot.com/2020/04/new-drawing-options-in-202003-release.htmlcolors = [(0.8,0.0,0.8),(0.8,0.8,0),(0,0.8,0.8),(0,0,0.8)]athighlights = defaultdict(list)arads = {}for i,rng inenumerate(rings.AtomRings()):for aid in rng: athighlights[aid].append(colors[i]) arads[aid] =0.3bndhighlights = defaultdict(list)for i,rng inenumerate(rings.BondRings()):for bid in rng: bndhighlights[bid].append(colors[i])
(From Wikipedia) The partition coefficient, abbreviated P, is defined as the ratio of the concentrations of a solute between two immisible solvents at equilibrium. Most commonly, one of the solvents is water, while the second is hydrophobic, such as 1-octanol.
Hence the partition coefficient measures how hydrophilic (“water-loving”) or hydrophobic (“water-fearing”) a chemical substance is. Partition coefficients are useful in estimating the distribution of drugs within the body. Hydrophobic drugs with high octanol-water partition coefficients are mainly distributed to hydrophobic areas such as lipid bilayers of cells. Conversely, hydrophilic drugs (low octanol/water partition coefficients) are found primarily in aqueous regions such as blood serum.
The dataset used in this notebook is obtained from Kaggle. This dataset features relatively simple molecules along with their LogP value. This is a synthetic dataset created using XLogP and does not contain experimental validation.
Let’s try building a model to predict a molecule’s logP value given other descriptors. We will try simple molecular descriptors and check the performance. Some molecule discriptors we will consider: 1. Molecular weight 2. Number of rotatable bonds 3. Number of aromatic compounds
logP_df.head(4)
SMILES
LogP
0
C[C@H]([C@@H](C)Cl)Cl
2.3
1
C(C=CBr)N
0.3
2
CCC(CO)Br
1.3
3
[13CH3][13CH2][13CH2][13CH2][13CH2][13CH2]O
2.0
As before we will first convert the SMILES string into a rdkit.Chem.rdchem.Mol object, let’s write a convenience function to do so
# Getting count of aromatic elements _count =0for i inrange(mol.GetNumAtoms()):if mol.GetAtomWithIdx(i).GetIsAromatic(): _count = _count +1print(_count)
/var/folders/_r/6hhq_8ps5118p6sqqz231j480000gq/T/ipykernel_26421/2485801931.py:2: UserWarning: To output multiple subplots, the figure containing the passed axes is being cleared
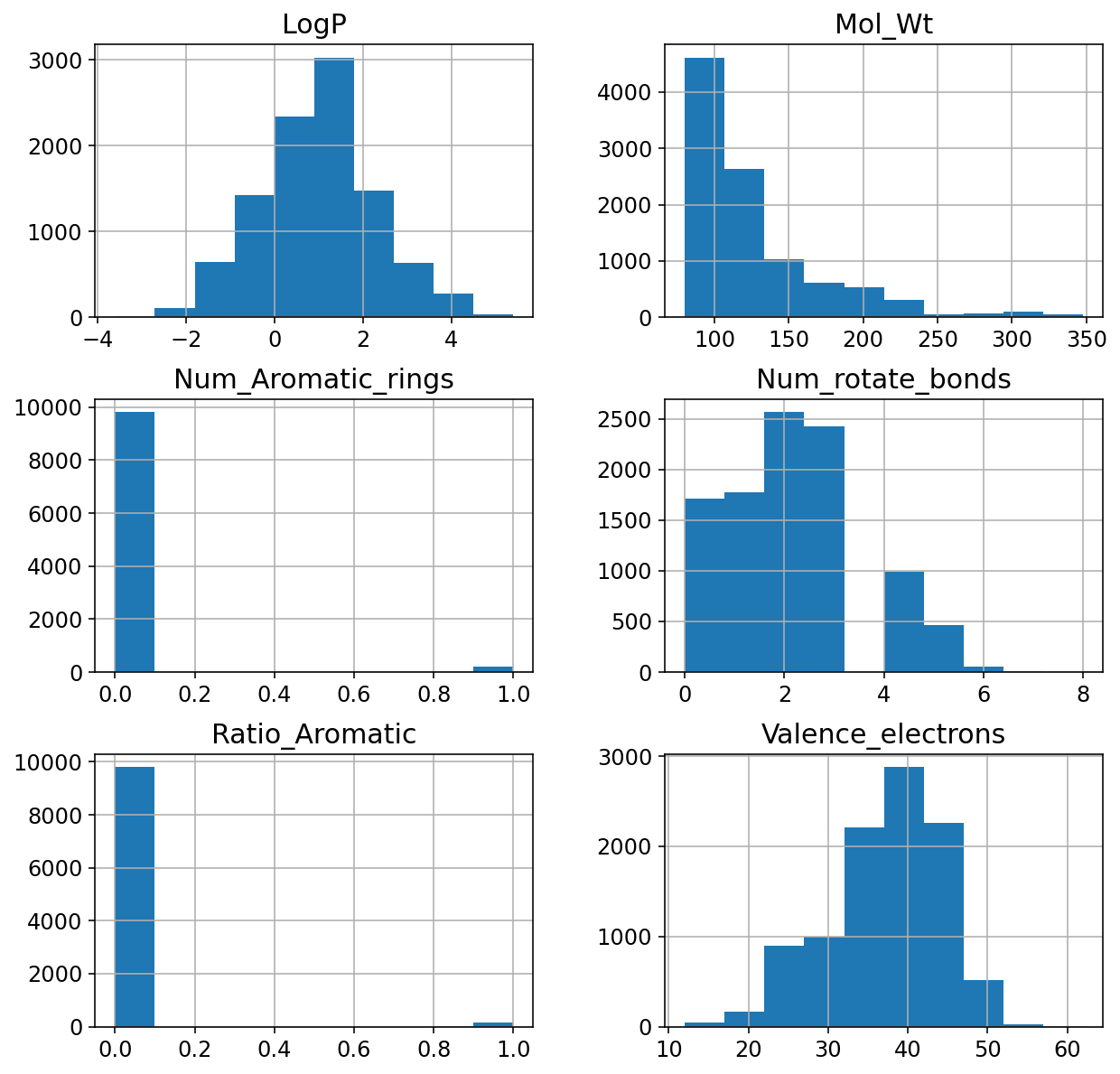
df_var_10k.hist(ax = ax);
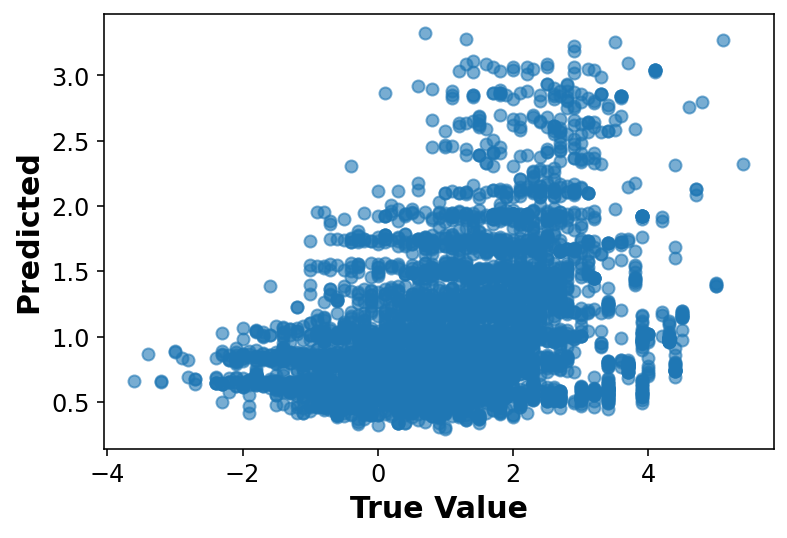
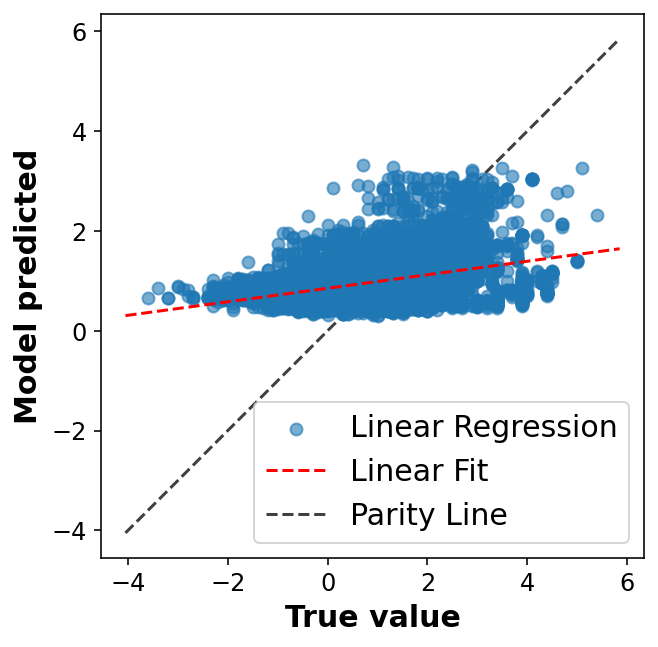
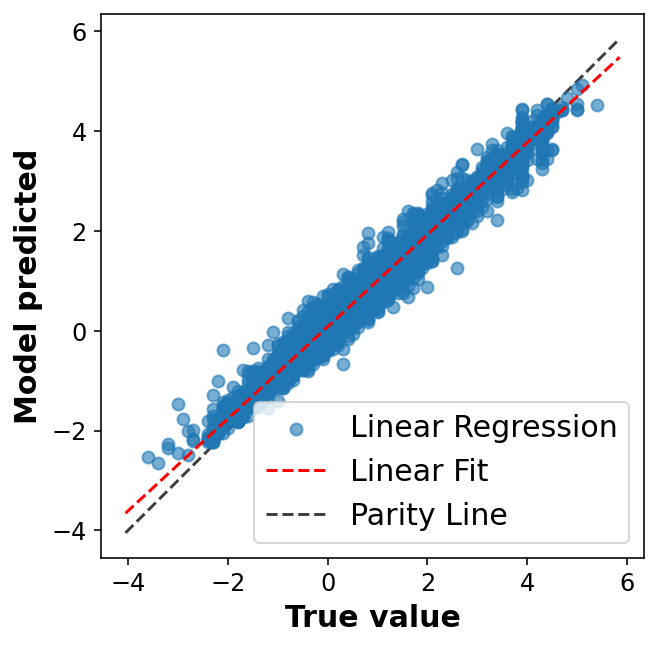
# Fancier looking plot fig, ax = plt.subplots(1,1, figsize=(5,5))ax.scatter(y_train, y_pred, alpha=0.6, label='Linear Regression')lims = [np.min([ax.get_xlim(), ax.get_ylim()]), # min of both axes np.max([ax.get_xlim(), ax.get_ylim()]), # max of both axes ]# Linear fitreg = np.polyfit(y_train, y_pred, deg=1)ax.plot(lims, reg[0] * np.array(lims) + reg[1], 'r--', linewidth=1.5, label='Linear Fit')ax.plot(lims, lims, 'k--', alpha=0.75, zorder=0, label='Parity Line')ax.set_aspect('equal')ax.set_xlabel('True value')ax.set_ylabel('Model predicted')ax.legend(loc='best');
Evaluate model success
One of the simplest ways we can evaluate the success of a linear model is using the coefficient of determination (R2) which compares the variation in y alone to the variation remaining after we fit a model. Another way of thinking about it is comparing our fit line with the model that just predicts the mean of y for any value of x.
Another common practice is to look at a plot of the residuals to evaluate our ansatz that the errors were normally distributed.
In practice, now that we have seen some results, we should move on to try and improve the model. We won’t do that here, but know that your work isn’t done after your first model (especially one as cruddy as this one). It’s only just begun!
Compress molecules into vectors for mathetical operations and comparisons. First we will look at MorganFingerprint method. For this method we have to define the radius and the size of the vector being used.
More information on different Circular Fingerprints can be read at this blogpost. Highly recommended
radius =2# How far from the center node should we look at? ecfp_power =10# Size of the fingerprint vectors ECFP = [ np.array(AllChem.GetMorganFingerprintAsBitVect(m, radius, nBits =2**ecfp_power)) for m in df_train['ROMol'] ]
# Standard Scalingstd_scaler = StandardScaler()X_train_std = std_scaler.fit_transform(X_train)
model = RandomForestRegressor()model.fit(X_train_std, y_train)
RandomForestRegressor()
y_pred = model.predict(X_train_std)
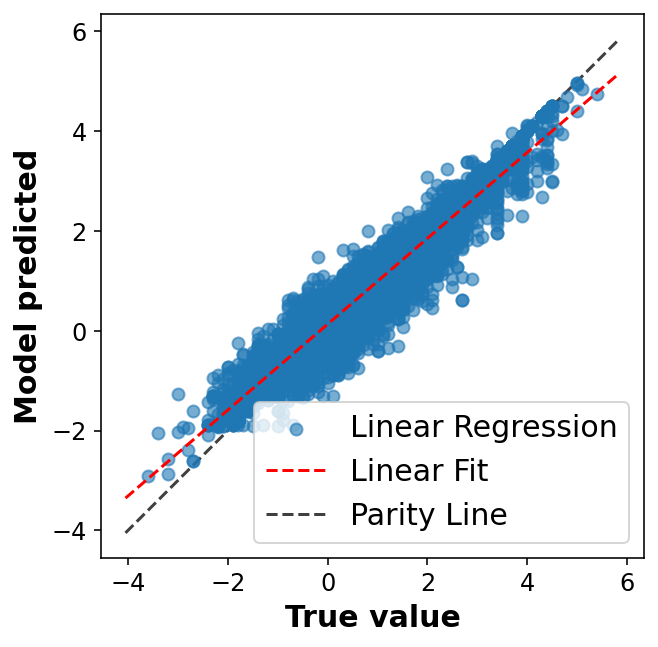
# Fancier looking plot fig, ax = plt.subplots(1,1, figsize=(5,5))ax.scatter(y_train, y_pred, alpha=0.6, label='Linear Regression')lims = [np.min([ax.get_xlim(), ax.get_ylim()]), # min of both axes np.max([ax.get_xlim(), ax.get_ylim()]), # max of both axes ]# Linear fitreg = np.polyfit(y_train, y_pred, deg=1)ax.plot(lims, reg[0] * np.array(lims) + reg[1], 'r--', linewidth=1.5, label='Linear Fit')ax.plot(lims, lims, 'k--', alpha=0.75, zorder=0, label='Parity Line')ax.set_aspect('equal')ax.set_xlabel('True value')ax.set_ylabel('Model predicted')ax.legend(loc='best');
display_performance(y_train,y_pred);
R2: 0.97
MAE: 0.15
RMSE: 0.22
Similarity
RDKit provides tools for different kinds of similarity search, including Tanimoto, Dice, Cosine, Sokal, Russel… and more. Tanimoto is a very widely use similarity search metric because it incorporates substructure matching. Here is an example
ref_mol = df_var_10k.iloc[4234]['ROMol']
ref_mol
# Generate finger print based representation for that molecule ref_ECFP4_fps = AllChem.GetMorganFingerprintAsBitVect(ref_mol, radius=2)
df_var_10k_ECFP4_fps = [AllChem.GetMorganFingerprintAsBitVect(x,2) for x in df_var_10k['ROMol']]
Estimate the similarity of the molecules in the dataset to the reference dataset, there are multiple ways of doing it: we are using Tanimoto fingerprints
from rdkit import DataStructssimilarity_efcp4 = [DataStructs.FingerprintSimilarity(ref_ECFP4_fps, x) for x in df_var_10k_ECFP4_fps]